¶ Key elements of neurogenic inflammation in migraine (Ramachandran, 2018)
The etiology of migraine pain involves sensitized meningeal afferents that densely innervate the dural vasculature.
Factors such as chronic stress, diet, hormonal fluctuations, or events like cortical spreading depression can generate a state of sterile inflammation in the intracranial meninges, activating meningeal nociceptors.
The cell bodies of the afferent nerves are located in the trigeminal ganglion, project to the nucleus caudalis, and sends information to higher brain centers.
There is a release of neuropeptides (such as substance P, calcitonin gene related peptide) from the trigeminal innervation leading to vasodilation, plasma extravasation secondary to capillary leakage, edema, and mast cell degranulation.
Ramachandran R. Neurogenic inflammation and its role in migraine. Semin Immunopathol. 2018 May;40(3):301-314. doi: 10.1007/s00281-018-0676-y. Epub 2018 Mar 22. PMID: 29568973.
Peroutka SJ. Neurogenic inflammation and migraine: implications for the therapeutics. Mol Interv. 2005 Oct;5(5):304-11. doi: 10.1124/mi.5.5.10. PMID: 16249526.
Costa C, Tozzi A, Rainero I, Cupini LM, Calabresi P, Ayata C, Sarchielli P. Cortical spreading depression as a target for anti-migraine agents. J Headache Pain. 2013 Jul 23;14(1):62. doi: 10.1186/1129-2377-14-62. PMID: 23879550; PMCID: PMC3728002.
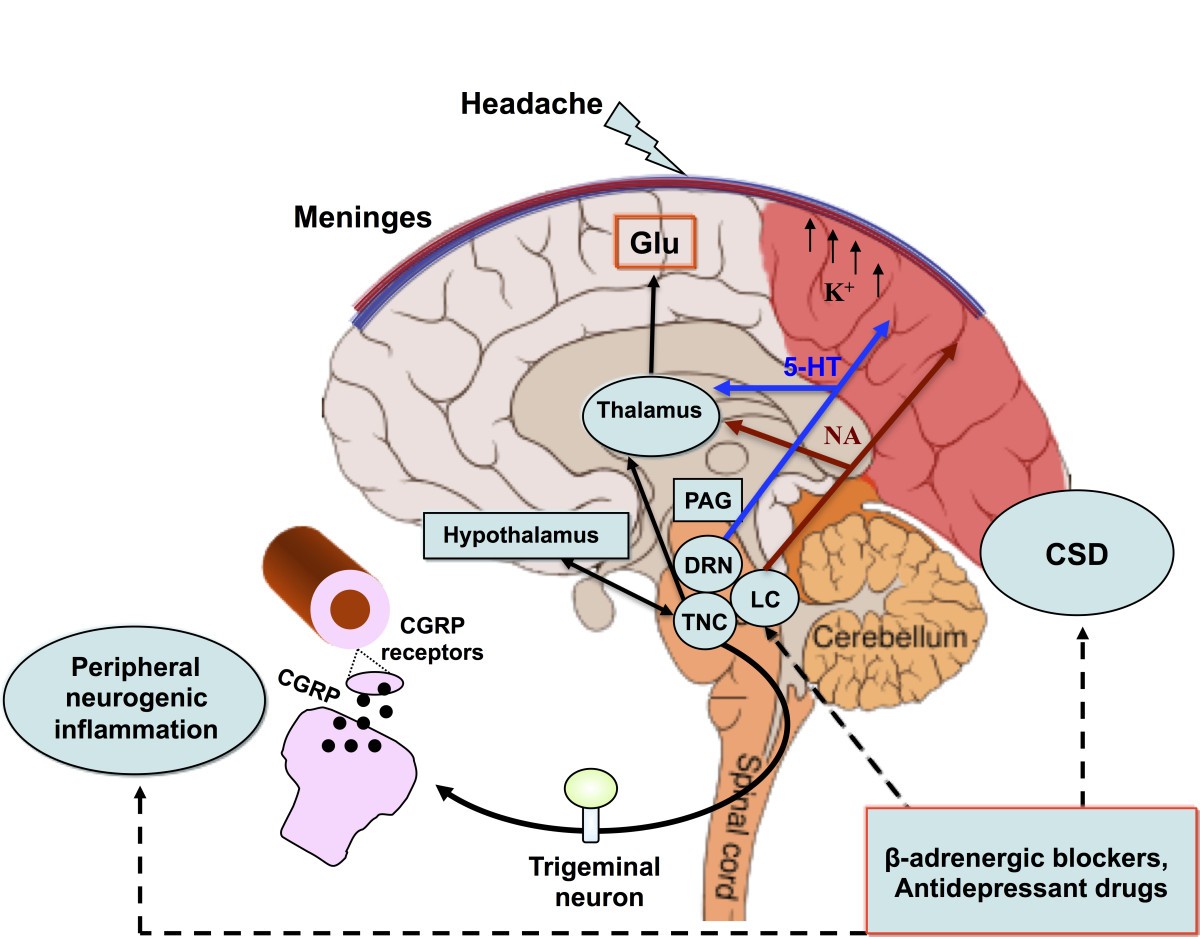
Mechanisms and structures involved in the pathogenesis of migraine with aura. CSD underlies aura symptoms. Initiation and propagation of CSD are determined by massive increases in extracellular potassium ion concentration and excitatory glutamate. CSD biochemical changes may trigger the activations of meningeal trigeminal endings and trigemino-vascular system, causing the headache phase. The latter can occur through matrix metalloproteases activation that increases vascular permeability and also through the release of nociceptive molecules from mastcells, including proinflammatory cytokines. The pain phase is due to peripheral and central sensitization of the trigeminal system, as well as to the release of CGRP, both peripherally and centrally. CGRP is considered a key mediator in migraine and, together with NO, is the main molecule responsible for vasodilation consequential to neurogenic inflammation. CGRP is also released from cortical slices during CSD and this calcium-dependent release can mediate the dilatation of cortical arterioles. Periacqueduttal gray matter (PAG), locus coeruleus (LC), nucleus of raphe magnum (NRM) are brainstem structures implicated in the processing of trigeminal pain. Functional and structural PAG abnormalities occurring in migraineurs, contribute to the hyper-excitability of trigeminal nociceptive pathways. Functional alteration of noradrenergic nuclei of the LC are believed to be involved in cortical vasomotor instability. Thus, dysfunction in brainstem pain-inhibiting circuitry may explain many facets of the headache phases, even in MwA. CSD participates in this dysregulation by antagonizing the suppressive effect exerted by NRM on the responses of trigeminal neurons. In this scenario, amitriptyline may influence CSD by preserving 5-HT and perhaps NA neurotransmission in the cortex and/or inhibiting high-voltage-activated (HVA) Ca2+ channels and Ca2+ currents. Propranolol, on the other hand, may reduce neuronal excitability and susceptibility to CSD via a beta-adrenergic blockage. Original brain template was designed by Patrick J. Linch, medical illustrator and C. Carl Jeffe, MD, cardiologist.